
-
- 15年项目操作经验
- 咨询服务
-
- 合作各知名品牌
-
- 大型医疗机构合作商
-
- 提供行业解决方案


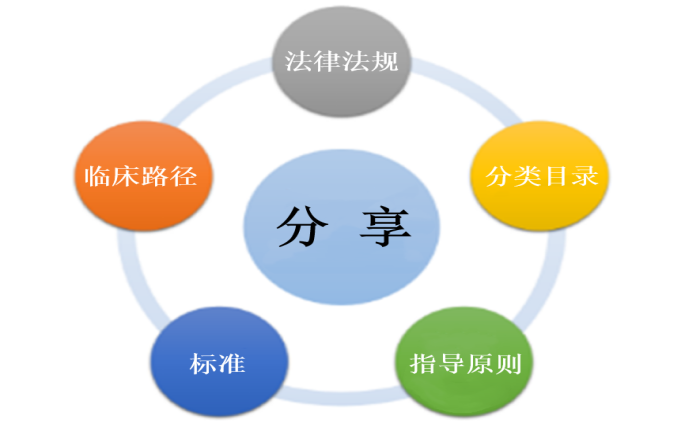
Pre Market Research Service
NMPA Registration/Recordation Consulting Service
CRO Service
International Certification Service
CDMO Service
Intellectual Property Service
Disinfection Product Recordation Service
威恩普斯合规咨询(Winpush)是一家致力于医疗器械全生命周期的合规咨询服务机构。我们为国内外医疗器械注册人及制造商提供产品上市过程中任意阶段的法规咨询服务。服务内容含盖:质量管理体系咨询及辅导、产品上市前研究辅导、NMPA注册/备案服务、产品检验代理服务、临床评价服务(同品种临床评价、临床试验CRO)、国际认证服务等...【详细】


APET/CT产品注册申报时,若其中的CT已取得注册证书,CT相关的综述资料、研究资料应如何提供?
Q 综述资料中应明确CT的制造商、型号、注册证书编号;同时说明PET/CT系统中的CT和已取得注册证书的CT有哪些差异。对差异进行分析,提交差异对安全性、有效性影响的研究资料。若CT部分和原注册证书没有差异,可提供PET/CT 系统的研究资料,不再单独提供CT部分的研究资料。(源于CMDE一部)
A脊柱后路非融合固定系统产品注册单元应如何划分?
Q 脊柱后路非融合固定系统用于脊柱后路非融合固定,与用于融合的脊柱后路内固定系统不同,应划分为不同的注册单元。产品的工作原理不同、结构设计不同,应分为不同的注册单元。产品起主要功能作用的材料不同,应划分为不同注册单元。若产品组成部件(如钉)材料不同,但作为整体组配或组合使用的产品可按同一注册单元申报。(源于CMDE四部)
A关于医用缝合针产品的首次注册以及许可事项变更中,同一注册单元内注册检验典型性产品的确定原则?
Q 按照“同一注册单元内,所检测的产品应当是能够代表本注册单元内其他产品安全性和有效性的典型产品”的原则,进行注册检验的典型性产品应能涵盖该注册单元全部产品特征。如同一注册单元中有不同针型、不同牌号不锈钢的缝合针,则应考虑分别进行注册检验。(源于CMDE五部)
A水胶体敷料临床豁免情况不包括哪些?
Q对应《医疗器械分类目录》(2017年第104号)中14-10-05中举例水胶体敷料或水胶体敷贴。豁免情况不包括: (1)适应症宣称可以促进上皮化、引导组织再生、促进伤口愈合、减轻疼痛、抗菌、防感染、抗病毒、止血、溶解坏死组织、减少疤痕、防粘连、作为人工皮/皮肤替代物等作用的产品; (2)宣称可以用于体内伤口、三度烧伤、感染创面、坏死组织较多的创面、发生创面脓毒症的患者等情况的产品; (3)含有活性成分的产品:如药品/药用活性成分、生物制品/生物活性成分、银、消毒剂等; (4)其他新型产品,如新材料、新作用机理、新功能的产品。(源于CMDE五部)
A栓塞微球产品生物学评价中的“致癌性”评价项目,是否可以通过“材料化学表征及安全性分析”来验证和评价?
Q 该类产品为植入器械,预期与循环血液持久接触(>30d)。申请人应参照现行GB/T16886.1 进行生物学评价,综合申报产品材料(包括可能残留的加工助剂)的境内外上市后临床应用情况进行评价。充分的化学表征及毒理学评价研究可为免于致癌试验提供支持。对于首次应用于植入性医疗器械的材料,现有毒理学评价研究可能不充分。(源于CMDE三部)
AECMO产品动物试验评价指标包括哪些?
Q 通常情况下,ECMO产品的动物试验中建议包括(但不限于)以下指标:血液相关的实验室检查指标;病理学相关指标;生命体征及血流动力学监测指标;ECMO运行期间监测并记录主要治疗参数,观察器械外观变化,监测可能的器械故障;定期查看实验动物插管部位是否有机械损伤和感染迹象;动物试验中,宜对任何安全性事件/异常进行记录和分析;实验动物尸检时,应同时观察器械外观变化,也可以对回收器械进行体外性能检测。 若需验证申报产品的某些特殊功能和/或性能时,可选择针对相关功能和/或性能的特定评价指标,同时提供相关评价指标的确定依据。(源于CMDE五部)
A如何开展药物洗脱支架的细胞毒性评价?
Q支架部分和输送系统应分别开展细胞毒性评价。如含药支架部分细胞毒性较高,应进行原因分析,并进行综合评价。例如,细胞毒性考虑由药物引起时,应分别开展裸支架及含药支架的细胞毒性试验,并对含药支架细胞毒性进行风险分析,综合评价所含药物的影响。(源于CMDE三部)
A与循环血液接触的医疗器械,是否将热原和细菌内毒素均列入产品技术要求?
Q 与循环血液接触的医疗器械,依据《医疗器械产品技术要求编写指导原则》中要求,细菌内毒素需在技术要求中制定。热原项目可在生物相容性研究资料中提供。产品注册时,细菌内毒素和热原均需进行研究。(源于CMDE三部)
A产品按二类注册申报时已提交过检测报告,在产品管理类别调整为三类后,按照三类注册申报时是否可使用原检测报告?
Q 检验报告没有有效期。如果产品未发生任何变化,可以提交原检验报告。(源于CMDE二部)
A有源医疗器械中可重复使用的附件消毒灭菌资料应关注的问题有哪些?
Q 可重复使用的附件,使用前应保证已消毒或灭菌。说明书中应明确具体的消毒/灭菌方法(如使用的消毒剂、消毒或灭菌设备),消毒/灭菌周期的重要参数(如时间、温度和压力等)。研究资料中应提供消毒/灭菌方法确定的依据、消毒/灭菌效果确认资料及推荐的消毒/灭菌方法耐受性的研究资料。(源于CMDE一部)

服务热线:0531-59525938
0531-59525939
联系邮箱:info@win-push.com
公司地址:山东省济南市历下区茂陵山路2号普利商务中心
 公众号
公众号
 微信客服
微信客服
微信客服






